心筋細胞における核内Ca2+濃度調節の重要性:刺激に応じた制御と生理的役割
Tweet執筆者情報
執筆者:中村(西谷)友重1、中尾周1、若林繁夫1,2
執筆者所属:1国立循環器病研究センター研究所・分子生理部、2同研究所・心臓生理機能部
原著論文:Stimulus-Dependent Regulation of Nuclear Ca2+ Signaling in Cardiomyocytes: A Role of Neuronal Calcium Sensor-1. (PLoS ONE 10:e0125050, 2015)
更新日:2015年7月8日

概要
心臓の細胞内Ca2+は筋収縮や遺伝子発現調節のキー因子である。近年、細胞質のみならず核内Ca2+濃度調節の重要性が示唆されているが、詳細な制御機構や生理機能は不明である。本研究では、活動電位などの電気刺激およびホルモンなどによる受容体刺激により核内Ca2+濃度の増減が生じるが、異なるCa2+放出チャネルや制御因子を介して、異なる生理機能に寄与している可能性が示された。このことは、核が複雑な細胞内Ca2+シグナルを局所で制御する場としても働くという新規の機能を示唆している。
背景および方法
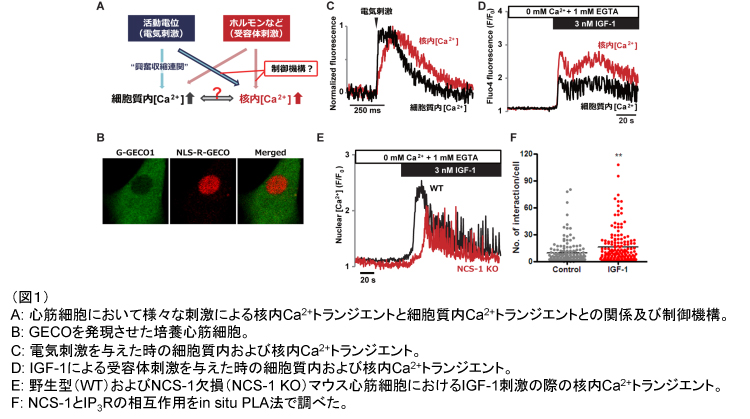
心臓において、細胞内Ca2+は筋収縮や遺伝子発現、細胞死など様々な細胞機能の調節因子として働いている。例えば、活動電位刺激により電位依存性Ca2+チャネルが活性化されると、細胞内Ca2+貯蔵装置といわれる筋小胞体(SR)から、細胞内Ca2+放出チャネルであるリアノジン受容体を介して大量のCa2+が細胞質内に放出され、これが筋収縮の引き金となる[1]。一方、ホルモンなどによる受容体刺激により、シグナル分子であるイノシトール三リン酸(IP3)が生成され、別の細胞内Ca2+放出チャネルであるIP3受容体を活性化して細胞内Ca2+濃度が上昇し、心肥大などの遺伝子発現に寄与する[2]。このように重要な働きを持つ細胞内Ca2+濃度の増減(Ca2+トランジエント)は、細胞質のみならず核内でも生じていることが近年報告されている[3]。しかし、1)核内Ca2+濃度が、拍動に伴って絶え間なく増減する細胞質内Ca2+とどのように区別されて制御されているのか、2)特に電気刺激や受容体刺激といった異なる刺激に対し、異なるメカニズムが存在するのか、3)核内Ca2+濃度上昇の生理的意義は何か、などについてはほとんどわかっていない(図1A参照)。
そこで本研究ではこれらを明らかにすることを目的に、電気刺激または受容体刺激を与えた時の核内および細胞質内Ca2+濃度を比較し、関連経路を解析した。同一細胞における核内および細胞質内Ca2+トランジエントを同時測定するため、部位特異的に発現する新規Ca2+感受性蛍光タンパク質GECO(Genetically Encoded Fluorescent Ca2+ Indicator)[4]と従来から使用されてきた蛍光Ca2+指示薬Fluo-4/AMを培養心筋細胞に導入し、それぞれ定性的および定量的に解析を行った。また、IP3受容体の機能を制御することが知られているNCS-1(Neuronal Ca2+ Senor-1)タンパク質の核内Ca2+制御における役割についても、遺伝子欠損マウス(KOあるいはNCS1-/-)を用いて検討した。
結果
■ 電気刺激を与えた時の核内および細胞質内Ca2+トランジエント
培養心筋細胞に細胞質特異的に発現するG-GECO1と核特異的に発現するNLS-R-GECOを導入すると、細胞質は緑色、核は赤色の蛍光が同一心筋細胞で観察された(図1B)。これらの細胞に電気刺激を与えると、細胞質内Ca2+レベルの増減に同期して収縮タンパク質の存在しない核内でもCa2+レベルが増減した。同様の結果が、Fluo-4/AMを用いた場合でも認められた。しかしLine-scanを用いた高速測定の結果、核内Ca2+動態は細胞質内Ca2+動態よりも遅延しており(図1C)、さらにCa2+結合タンパク質Parvalbumin[5]で細胞質内Ca2+を除去すると核内Ca2+レベルの上昇が抑えられた。すなわち、電気刺激により増加した核内Ca2+は細胞質由来であると考えられる。また、細胞内Ca2+放出チャネルの阻害剤を用いた実験結果から、電気刺激の際の核内Ca2+濃度上昇にはリアノジン受容体が寄与していることがわかった。
■ 受容体刺激を与えた時の核内および細胞質内Ca2+トランジエント
GECOとFluo-4/AM両方の系を用いてインシュリン様増殖因子IGF-1による受容体刺激を与えた時にも、核内および細胞質内両方においてCa2+濃度が上昇した。しかし、上昇したCa2+濃度の振幅は核内の方が高かった(図1D)。また、阻害剤の実験からIGF-1刺激による核内Ca2+濃度上昇にはIP3受容体が働いていることがわかった。このことは、電気刺激と受容体刺激では異なるメカニズムで核内Ca2+濃度上昇が生じることを示している。
■ 細胞内Ca2+センサーNCS-1の寄与
私達は以前、ホルモンなどの受容体刺激の際にはIP3受容体が活性化されて心肥大などが導かれるが、その際、NCS-1というCa2+センサータンパク質とIP3受容体との相互作用が重要であることを見出した[6]。NCS-1は主に神経などの興奮性細胞に高発現し、様々なイオンチャネル[7]やイノシトールリン酸キナーゼ(PI4K)などを活性化することにより神経機能に重要な役割を担う[8]ことが知られている、小さなCa2+結合タンパク質(22 kDa)である[9]。NCS-1は心肥大の際に発現上昇が認められたことから[6]、受容体刺激を与えた時の核内Ca2+濃度調節への関連についてKOマウスを用いて検討した。その結果、KOマウス由来の心筋細胞では、IGF-1刺激による核内および細胞質内Ca2+レベルの上昇が顕著に抑制されていた(図1E)。また、NCS-1とIP3受容体は心筋細胞の核膜周辺およびSRに共局在しており、さらにIGF-1刺激によってNCS-1とIP3受容体との相互作用が強化されることがわかった(図1F)。
考察
■ 新規Ca2+感受性蛍光タンパク質GECOによる測定
本研究は、細胞質または核に特異的に発現するGECOを用いて心筋細胞内で部位特異的にCa2+トランジエントを測定した初めての例である。期待通り、同一細胞において細胞質内および核内Ca2+濃度の同時測定ができた。従来の蛍光Ca2+プローブと較べ、細胞質内と核内Ca2+がオーバーラップして映るなどのアーチファクトがない点で優れている。一方で、部位特異的に使用する蛍光タンパク質が異なるため、核内と細胞質内Ca2+の濃度比較などの定量的な比較はできない。また、電気刺激によるCa2+トランジエントのように非常に速い反応では、GECOのシグナル変化量とCa2+との解離速度が低いためか高分解能の測定ができない。従ってGECOでは定性的な解析が、またFluo-4/AMでは定量的な解析が適していることがわかった。
■ 電気刺激と受容体刺激における核内Ca2+制御の違い
Ca2+濃度変動の時間経過及びParvalbuminの実験から、電気刺激による核内Ca2+トランジエントのCa2+供給源は主に細胞質であること、すなわち電気刺激の際にはまず細胞質内Ca2+濃度の上昇がリアノジン受容体を介して生じ、そのCa2+が(おそらく核膜孔を介した受動拡散により)核内へ移行して核内Ca2+濃度が上昇するのではないかと予測された。核が拍動に伴って増減する細胞質内Ca2+のバッファー的役割を担っているのかもしれない。一方、受容体刺激の場合は細胞質内より核内Ca2+上昇の方が優勢であったことから、主なCa2+供給源が核膜内である可能性が高い。
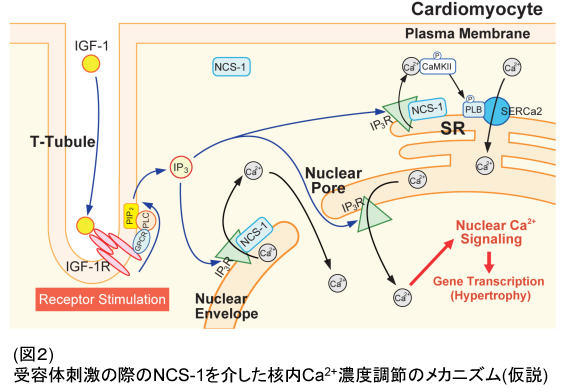
■ 受容体刺激の際のNCS-1を介した核内Ca2+濃度調節のメカニズム(仮説)
IGF-1刺激を与えるとNCS-1とIP3受容体の相互作用が強まり、核内及び細胞質内Ca2+レベル上昇に寄与するという結果が得られた。そのメカニズムとして以下の3つの仮説が考えられる。1)NCS-1が核の内膜に存在するIP3受容体を直接活性化することにより、核膜内を介して核内へのCa2+放出が増えた。2)核の外膜にあるIP3受容体の活性化を介して核周辺に放出されたCa2+が核膜孔を通って核内に流入した。3)NCS-1が核膜内のCa2+量を維持することにより、間接的に核膜内から核内へのCa2+放出が増えた(図2参照)。
このうち、1)に関してはNCS-1が核内にほとんど存在しないという私達の実験結果(データ示さず)から、可能性は低い。2)に関しては、NCS-1が核の外膜の周りでIP3受容体と共局在しており、Ca2+の拡散速度は細胞質内より核内の方が高いため、可能性は高い。3)に関しては、私達は以前、NCS-1がSRのCa2+量維持に寄与することを報告しており[6]、またSR内と核膜内は繋がっているため[10]、核膜内のCa2+量維持にもNCS-1が関与している可能性が高い。どの経路が最も関与しているのか今後調べる必要性がある。
まとめ
本研究により、電気刺激および受容体刺激どちらの場合でも核内Ca2+濃度は上昇するが、そのメカニズムや生理機能および関与する制御因子は異なるものと考えられる。このことは、核が遺伝子発現調節の場としてのみならずCa2+シグナル制御の場としても重要であることを示唆している。
参考文献
1. Bers, D. M. Nature 415, 198-205 (2002).
2. Wu, X. et al. J. Clin. Invest. 116, 675-682 (2006).
3. Ljubojevic, S. et al. Biophys. J. 100, 2356-2366 (2011).
4. Zhao, Y. et al. Science 333, 1888-1891 (2011).
5. Ibarra, C. et al. Circ. Res. 112, 236-245 (2013).
6. Nakamura, T. Y. et al. Circ. Res. 109, 512-523 (2011).
7. Nakamura, T. Y. et al. Proc. Natl. Acad. Sci. U. S. A. 98, 12808-12813 (2001).
8. Nakamura, T. Y. et al. J. Cell Biol. 172, 1081-1091 (2006).
9. Nakamura, T. Y. et al. Trends in Cell & Molecular Biology 7, 99-110 (2012).
10. Wu, X. et al. Circ. Res. 99, 283-291 (2006).