膵発癌モデルマウスを用いたゲムシタビンとエルロチニブの併用効果とメカニズムの検討
Tweet執筆者情報
執筆者:宮林弘至・伊地知秀明・小池和彦
執筆者所属:東京大学医学部付属病院消化器内科
原著論文:Erlotinib Prolongs Survival in Pancreatic Cancer by Blocking Gemcitabine-Induced MAPK Signals. Cancer Research 73:2221-2234, 2013.
更新日:2013年6月10日

概要
ヒト切除不能膵癌に対してゲムシタビンとEGFR阻害剤エルロチニブの併用療法はゲムシタビン単独と比較して有意に生存期間を延長したが、EGFRの下流のKRAS変異が90%以上に存在する膵癌におけるエルロチニブの効果の詳細は不明である。本研究で恒常性KrasG12D発現+TGF-betaⅡ型受容体ノックアウト膵発癌モデルを用いて、その作用機序としてゲムシタビン投与群でみられるEGFR/ERBB2とその下流のMAPKシグナルの活性化をエルロチニブが抑制することが示された。本モデルはヒト膵癌の臨床・組織像をよく近似し、新規薬剤の効果、作用機序の解析に有用であり、また治療の奏功する患者群の検討や治療抵抗性の機序解明にも有用と考えられた。
はじめに
膵癌は依然として最難治癌であり、様々な治療法に強い抵抗性を示す。膵癌の病態の理解に基づいた、より有効な治療法の開発は急務といえる。最近では分子標的薬を組み合わせて予後を延長する検討がなされており、これまでの数々の検討の中で、ゲムシタビンとEGFR阻害剤エルロチニブの併用はゲムシタビン単独と比較して、分子標的薬では唯一、統計学的有意に生存期間を延長した(1)。しかし、膵癌におけるEGFR阻害剤の効果の予測因子はいまだに解明されておらず、過去の報告でEGFR阻害剤が膵癌に効果を示すメカニズムを明らかにしたものはない。その原因として膵癌の大多数がEGFRの下流のKRASの活性型変異を有していること(2,3)、非小細胞肺癌で効果予測因子とされるEGFRの活性型変異が稀であることが挙げられる。また、膵癌では過去にEGFRの発現は全体の30-70%程度といわれているが、EGFR発現状態とEGFR阻害剤の効果との関連は報告されていない(1,4)。
本邦でエルロチニブが保険適応となり、生存期間の延長はわずかであるが、膵癌においてゲムシタビン単独に対して唯一統計学的有意に生存期間を延長したレジメンであるという臨床的なインパクトから、このレジメンは膵癌治療における重要な選択肢と考えられる。それゆえ、エルロチニブがゲムシタビンとの併用によってなぜ膵癌に効果を示すのか、そのメカニズムを解明することは、臨床的にも非常に重要であると考える。
前臨床的検討としてのマウスモデル
ヒト膵癌において、正常の膵臓上皮にKRAS、SMAD4などの遺伝子異常が蓄積するにつれ、前癌病変のPanIN (pancreatic intraepithelial neoplasia)が出現し、その段階が進行し、浸潤癌となるという多段階発癌仮説が臨床的にコンセンサスを得ている(図1)(5)。膵癌ではKRAS遺伝子の異常が90%以上とされ、膵癌の発癌に必須といえる。一方、癌抑制遺伝子とされるp16、p53、SMAD4遺伝子の異常・機能喪失も段階的に認められ、発癌プロセスの進行に寄与すると考えられる。その中でSMAD4の欠失・変異が50%以上と他の癌種に比べ高頻度に認められ、KRAS遺伝子とともにこれらのシグナルの異常が膵癌に特徴的に強く関わっていることが示唆される。近年、これら膵癌で臨床的にみられる遺伝子異常やシグナルの異常をマウスの膵臓に導入することで、ヒト膵癌類似の病変を呈するモデルが進歩してきた(6)。最近の報告では、遺伝子改変マウスモデルが移植モデルよりも癌の微小環境をより臨床像に忠実に再現すると考えられ、またヒトの臨床試験の生存効果もよりよく反映すると考えられるようになっている(7,8)。
われわれは、これまでにヒト膵癌のモデルとなる恒常性KrasG12D発現+TGF-betaⅡ型受容体(Tgfbr2)ノックアウトモデルを報告してきた(6)。このモデルは全例で急速に膵癌を形成し、腹部膨満、体重減少、腹水貯留、黄疸などのヒト膵癌類似の症候を呈する。その組織型は、豊富な間質を伴い線維化が著明な分化型管状腺癌であり、それまでの既報のマウスモデルで報告されていたヒトでは稀な肉腫様の腫瘍や、未分化癌がみられないという点で、最もヒト膵癌に近いモデルといえる (6)。
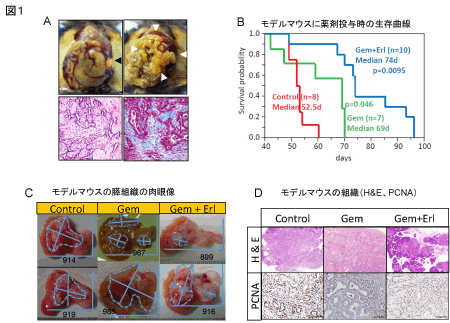
図1A Kras+Tgfbr2KOモデルの開腹所見と組織像
腹部は膨満し血性腹水の貯留あり、黄疸も認める(左上)。膵全体が腫瘍化し腫大・硬化している(右上)。膵組織はductal adenocarcinomaで(左下)、著名な線維化を伴う(右下、collagen:青)。
図1B Kras+Tgfbr2KOモデルの薬剤投与時の生存曲線
MST=GEM群69日vs無治療群52.5日、p=0.046、MST=GEM+erlotinib群74日vsGEM群69日、p=0.0095
図1C Kras+Tgfbr2KOモデルの薬剤投与時の膵組織の肉眼像
7週後の膵組織の肉眼像。青線で囲われているのが腫瘍部分。無治療群では膵癌組織は膵全体を占拠しており、GEM単独群で増殖が抑制されている個体がみられ、GEM+erlotinib併用群ではさらに増殖が抑制されていた。
Kras+Tgfbr2KOモデルおけるエルロチニブの効果
このKras+Tgfbr2KOモデルに膵癌の標準治療薬であるゲムシタビンとEGFR阻害剤のエルロチニブを投与した結果、ゲムシタビン単独投与でもKras+Tgfbr2KOマウスの生存期間は有意に延長し(生存期間中央値=ゲムシタビン群69日vs無治療群52.5日、p=0.046)、エルロチニブを併用するとさらに生存期間が延長した(生存期間中央値=ゲムシタビン+エルロチニブ群74日vsゲムシタビン群69日、p=0.0095)。生後7週で膵発癌モデルの膵組織を回収して比較すると、無治療群で膵全体を占拠していた膵癌組織は、ゲムシタビン単独群で増殖が抑制され、エルロチニブ併用群ではさらに増殖が抑制されることが肉眼像でも組織像でも確認され、本膵発癌モデルマウスでもエルロチニブの併用効果が示された(10)。
Kras+Tgfbr2KOモデルにおけるエルロチニブの効果の分子メカニズム
次にKras+Tgfbr2KOモデルの膵癌組織を用いて、効果のメカニズムの解析を試みた。免疫染色で、PCNA染色を施行すると無治療群に比較して、ゲムシタビン投与群で増殖が抑えられ、エルロチニブ併用群では増殖がさらに抑制されていることが示されたが、面白いことに、もともと無治療群では強くリン酸化されているEgfr、Erkが、ゲムシタビン投与群ではさらに増強していた。そしてエルロチニブを加えることで、その活性化が抑制されていた。ゲムシタビンの投与で癌の細胞死が誘導されるが、細胞死からのエスケープとしてEGFR-MAPK経路が活性化していることが考えられ、エルロチニブを併用すると活性化したEGFR-MAPK経路をさらに抑えるために効果を発揮することが示唆された。膵癌組織検体を用いたウェスタンブロット法においても、同様の結果が再現された。In vivoの実験と同様にin vitroの実験でもゲムシタビンがERKのリン酸化を増強し、エルロチニブの併用によりその活性化が抑制されることが示された。以上から、ゲムシタビンはKRASの状態にかかわらず膵癌細胞に対してEGFR-MAPK経路の活性化を誘導し、そこにerlotinibを併用することにより、その活性化が抑制されることが示された(10)。
ゲムシタビンによるEGFR-MAPK経路活性化のメカニズム
ゲムシタビンによるEGFR-MAPK経路の活性化の機序を明らかにするため、ゲムシタビンによるEGFRリガンドの発現への影響をReal-time PCRとELISAを用いて検討した。ゲムシタビン投与により、マウス膵癌細胞株K375におけるReal-time PCRでは有意にTgf-α、Egfの発現レベルが上昇し、マウス膵癌組織検体におけるELISAでは有意にAmphiregulin、Egfの分泌が亢進することが示された。ゲムシタビンが膵癌細胞においてEGFRリガンドの発現・分泌を誘導することによりEGFR-MAPK経路を活性化することが示唆された(10)。 次に受容体型チロシンキナーゼ(RTK)リン酸化アレイを用いて、ゲムシタビンがEGFR以外のRTKに与える影響を検討した。特にEgfr高発現群では、ゲムシタビンの投与でEgfrリン酸化が増強しており、エルロチニブ併用投与群ではEgfrリン酸化が抑制されていた。興味深いことにゲムシタビン投与群ではErbb2のリン酸化がEgfrリン酸化より強く認められ、エルロチニブ併用群ではその活性化も抑制されていた。膵癌細胞株を用いたin vitroの検討でもゲムシタビン投与によりErbb2の発現・リン酸化が亢進し、エルロチニブ併用により抑制された。またゲムシタビンの投与によりEgfrとErbb2のheterodimer形成が促進され、エルロチニブ併用で抑制された(10)。 さらに、MEK阻害剤を用いてMAPK経路を抑制するとゲムシタビンによるEGFRのリン酸化亢進が誘導されないことがわかり、ゲムシタビンによるEGFR活性化にはMAPK経路の活性化が重要であることがわかった(10)。 以上の結果から、膵癌に対するゲムシタビンによるEGFR-MAPK経路活性化のメカニズムとして、EGFRリガンドの分泌亢進と、ERBB2の活性化が関与していると考えられ、その活性化にはMAPK経路の活性化が重要であることが考えられた。膵癌においては、KRAS変異が高率ではあるものの、その上流に位置するEGFR、ERBB2といったERBBファミリーが治療標的及び生体マーカーとして重要であることが示唆される。
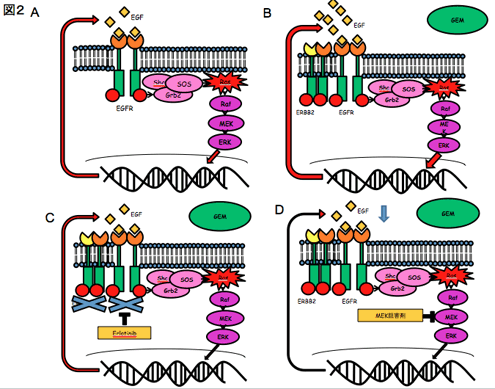
図2
A 膵癌においてKRAS変異によりMAPKシグナルが活性化している。
B ゲムシタビンの投与でEGFRリガンドの発現・分泌が亢進し、EGFR/ERBB2ヘテロダイマー形成が亢進し、EGFR-MAPKシグナルが活性化する。
C エルロチニブはゲムシタビンによるEGFR-MAPKシグナルの活性化を抑制する。
D ゲムシタビンによるEGFR-MAPKシグナル活性化にはMAPKシグナルが活性化していることが重要。
おわりに
Kras+Tgfbr2KOモデルは化学療法感受性もヒト膵癌に類似していると考えられ、治療薬の効果やメカニズムを解明するのに有用である。そして効果の予測因子・より効果のある治療薬の組み合わせ・より効果の得られる患者集団の特定など臨床的に重要な事項にも新たな知見を与えてくれる可能性がある。このモデルを用いて、KRAS変異が高率である膵癌におけるEGFR阻害剤の効果のメカニズムの一端を明らかにすることができた。ゲムシタビンがEGFR-MAPKシグナルを活性化し、KRAS変異のある膵癌においてもエルロチニブの併用でこの活性化が劇的に抑制された。このモデルを用いて今後の膵癌治療の発展につなげたい。
参考文献
1. Moore, M. J., Goldstein, D., Hamm, J., et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 25, 1960-6, (2007).
2. Almoguera C, Shibata D, Forrester K, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 53, 549-54, (1988).
3. Tada M, Omata M, Ohto M. Clinical application of ras gene mutation for diagnosis of pancreatic adenocarcinoma. Gastroenterology 100, 233-8, (1991).
4. da Cunha Santos G, Dhani N, Tu D, et al. Molecular predictors of outcome in a phase 3 study of gemcitabine and erlotinib therapy in patients with advanced pancreatic cancer: National Cancer Institute of Canada Clinical Trials Group Study PA.3. Cancer 16, 5599-5607, (2010).
5. Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer 2, 897-909, (2002).
6. Ijichi, H., Chytil, A., Gorska, A. E., et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev 20, 3147-60, (2006).
7. Olive, K. P., Jacobetz, M. A., Davidson, C. J., et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457-61, (2009).
8. Singh, M., Lima, A., Molina, R., et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat Biotechnol 28, 585-93, (2010).
9. Ijichi H, Chytil A, Gorska AE, et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest 121:4106-17, (2011)
10. Miyabayashi K, Ijichi H, Mohri D, et al. Erlotinib prolongs survival in pancreatic cancer by blocking gemcitabine-induced MAPK signals. Cancer Res 73, 2221-34, (2013).