Dock3は正常眼圧緑内障モデルマウスにおいてグルタミン酸毒性と酸化ストレスによる神経細胞死を抑制する
Tweet執筆者情報
執筆者:行方和彦、原田高幸
連絡先:原田高幸、〒156-8506 東京都世田谷区上北沢2-1-6 東京都医学総合研究所 視覚病態プロジェクトプロジェクトリーダー
原著論文:Dock3 attenuates neural cell death due to NMDA neurotoxicity and oxidative stress in a mouse model of normal tension glaucoma. Cell Death and Differentiation 20:1250-1256, 2013.
更新日:2014年1月10日

概要
緑内障は網膜神経節細胞死と視神経軸索の消失により視機能障害を引き起こす神経変性疾患であり、国内における最大の失明原因である。我々は神経特異的に発現するグアニンヌクレオチド交換因子であるDock3を網膜神経節細胞に過剰発現させると、神経細胞死が抑制されることを見出した。Dock3はグルタミン酸受容体の細胞内移行と分解を促進することによって、グルタミン酸毒性を低減すると考えられる。さらに緑内障モデルマウスにおいてもDock3による神経保護効果が認められた。
グルタミン酸受容体と神経毒性
グルタミン酸は中枢神経系における主要な神経伝達物質であり、学習や記憶はもちろん、視覚においても重要なはたらきをしている。通常、細胞外のグルタミン酸濃度はグルタミン酸輸送体などによって適切に制御されているが、虚血などの病的条件下では、過剰量のグルタミン酸による興奮毒性が神経細胞死を誘導する。グルタミン酸受容体はイオンチャネル型と代謝型に分類され、イオンチャネル型はさらに、N-methyl-D-aspartate(NMDA) 受容体と非NMDA受容体の2種類に分類される。NMDA受容体は電位依存的な活性調節を受けており、カルシウムイオンに対する高い透過性を有し、シナプス伝達効率や可塑性に影響を与えることが知られている。NMDA受容体のサブユニットには、NR1、NR2(NR2A、NR2B、NR2C、NR2Dの4種類)、NR3(NR3A、NR3Bの2種類)が知られている。NMDA受容体は通常、NR1の2量体とNR2またはNR3による4量体で構成されており、特にNR1/NR2Bで構成されたNMDA受容体は、神経細胞死に深く関与すると考えられている (1)。
NR2Bサブユニットと神経変性
NR2Bの細胞内ドメインにはAdaptor protein-2 (AP-2) およびPost synaptic dencity-95 (PSD95) と結合する領域が存在しており、これらがNR2Bを含むNMDA受容体のシナプス後膜での局在化を制御している。NR2Bの1472番目のアミノ酸であるTyrosine(Tyr1472)がFynキナーゼによりリン酸化されると、NR2BとAP-2との結合が阻害され、シナプス後膜からのエンドサイトーシスによる細胞内への取り込みが抑制される (2)。一方、PSD95との結合はTyr1472のリン酸化には依存せず、シナプス後膜に留まる方向に作用する。以上のような制御機構は、シナプス後膜に局在するNMDA受容体を増大させ、グルタミン酸への感受性を高めるためのメカニズムと考えられる。しかし最近になってこのメカニズムが、アルツハイマー病の増悪に関与することが報告された(3)。アミロイドβ毒性によって誘発される過剰リン酸化タウは、Fynキナーゼを樹状突起へ集積させて、NR2BのTyr1472リン酸化を促進する。したがって細胞膜におけるNR2Bの発現量が増加することにより、アミロイドβ毒性にグルタミン酸毒性が加わって、神経変性が増悪するものと考えられる。以上の結果はNR2Bのリン酸化と発現量の制御が、ある種の神経変性疾患における治療のターゲットとなり得ることを示唆している。
Dock3による神経保護効果
我々はNR2Bの細胞内ドメインが、Dock3と呼ばれるタンパクと結合することを見出した。Dock3はDock familyとよばれる分子群に属し、Rho family であるRac1を活性化するグアニンヌクレオチド交換因子として知られている (4)。我々が独自に作製したDock3過剰発現(Tg)マウスでは、Rac1を介したアクチン細胞骨格、およびGSK-3βを介した微小管重合の活性化により、切断後の視神経再生が促進することを報告している (5-7)。通常、野生型マウスの眼球に過剰量のグルタミン酸を投与すると、網膜神経節細胞が死滅して減少する。ところがDock3 Tgマウスでは、グルタミン酸投与後のNR2B発現量が野生型マウスと比較して有意に減少し、神経細胞死も抑制されていた。Dock3は細胞表面におけるNR2Bの発現量を低減することで、グルタミン酸毒性を抑制した可能性がある。実際にDock3 Tg マウス由来の培養網膜神経節細胞では、グルタミン酸負荷後のカルシウム流入と神経細胞死が抑制される (8)。また詳しいメカニズムは検討中であるが、Dock3 Tg マウス由来の培養網膜神経節細胞は、過酸化水素による細胞死についても耐性を発揮することが確認された。
Dock3による緑内障の治療研究
緑内障はわが国で最大の、そして世界でも第2位の失明原因である。最近の調査で緑内障は40歳以上の約5%に発症し、潜在患者数は500万人とも推定されている。また今後の高齢化社会においては、さらなる患者数の増加が危惧されている。本症には高眼圧(眼球内の水分の循環不全などにより眼球内圧が高まること、つまり眼球が硬くなること)を示すものと、眼圧が正常範囲内にある「正常眼圧緑内障」がある。前者では高眼圧が悪化要因と考えられているが、我が国においては全体の約7割が眼圧上昇を示さない「正常眼圧緑内障」で占められており、その発症原因は長年謎に包まれてきた。一方、近年では高眼圧に加えて、酸化ストレスやグルタミン酸毒性が、緑内障の原因として注目を集めている。このグルタミン酸濃度を調節する主要な機構がグルタミン酸輸送体だが、特に重要なのが網膜の特殊なグリア細胞であるMuller細胞に存在するGLASTである (9)。我々はGLAST欠損(KO)マウスでは眼圧が正常であるにも関わらず、グルタミン酸毒性と酸化ストレスの亢進により、網膜の神経細胞が加齢に伴い変性することを見出した。このマウスではヒト緑内障と同様の視神経の萎縮と視覚機能の障害も確認しており、世界初の正常眼圧緑内障モデルとして活用している (10)。
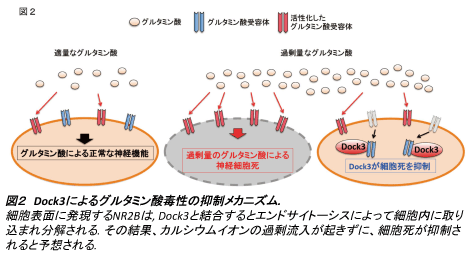
そこで次にGLAST KOマウスとDock3 Tgマウスを交配し、神経保護効果が得られるかどうかを検討した。その結果、GLAST KO:Dock3 Tgマウスでは、GLAST KOマウスで観察される緑内障の進行が抑制されることがわかった(図1)。さらにNR2BのTyr1472におけるリン酸化状態を調べたところ、GLAST KOマウスではリン酸化が亢進していたが、GLAST KO:Dock3 Tgマウスでは野生型マウスと同レベルまで低下していた。Dock3と結合したNR2BではTyr1472のリン酸化が抑制され、細胞内部への引き込みと分解が促進されたために、グルタミン毒性から保護されたことが考えられる(図2)。今後はDock3によるNR2B分解の制御機構について、さらなる解析を進めて行く予定である。

ヒトは外部環境からの情報の80%以上を視覚情報に頼っており、視機能の低下はQuality of Lifeの点からも深刻な事態を引き起こす。将来的に遺伝子治療等によって網膜神経節細胞にDock3を発現させる手法が確立すれば、緑内障などの治療法開発にも応用できるかもしれない。
参考文献
1. Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 14:383-400, 2013.
2. Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 47:845-857, 2005.
3. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 142:387-397, 2010.
4. Cote JF, Vuori K. GEF what? Dock180 and related proteins help Rac to polarize cells in new ways. Trends Cell Biol 17:383-393, 2007.
5. Namekata K, Harada C, Taya C, Guo X, Kimura H, Parada LF, Harada T. Dock3 induces axonal outgrowth by stimulating membrane recruitment of the WAVE complex. Proc Natl Acad Sci USA 107:7586-7591, 2010.
6. Namekata K, Watanabe H, Guo X, Kittaka D, Kawamura K, Kimura A, Harada C, Harada T. Dock3 regulates BDNF-TrkB signaling for neurite outgrowth by forming a ternary complex with Elmo and RhoG. Genes Cells. 17:688-697, 2012.
7. Namekata K, Harada C, Guo X, Kimura A, Kittaka D, Watanabe H, Harada, T. Dock3 stimulates axonal outgrowth via GSK-3β-mediated microtubule assembly. J Neurosci 32:264-274, 2012.
8. Bai N, Hayashi H, Aida T, Namekata K, Harada T, Mishina M, Tanaka K. Dock3 interaction with a glutamate-receptor NR2D subunit protects neurons from excitotoxicity. Mol Brain. 6:22, 2013.
9. Harada T, Harada C, Watanabe M, Inoue Y, Sakagawa T, Nakayama N, Sasaki S, Okuyama S, Watase K, Wada K, Tanaka K. Functions of the two glutamate transporters GLAST and GLT-1 in the retina. Proc Natl Acad Sci U S A. 95:4663-6, 1998.
10. Harada T, Harada C, Nakamura K, Quah HM, Okumura A, Namekata K, Saeki T, Aihara M, Yoshida H, Mitani A, Tanaka K. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J Clin Invest 117:1763-1770, 2007.