Jak2-V617FとLynからのGSK3およびmTOR経路を介した生存と増殖のシグナル伝達機構の新規樹立Jak2-V617F陽性AML細胞株PVTL-1における解析
Tweet執筆者情報
執筆者:長尾俊景、三浦修
執筆者所属:東京医科歯科大学医学部血液内科
原著論文:Proliferation and Survival Signaling from Both Jak2-V617F and Lyn Involving GSK3 and mTOR/p70S6K/4EBP1 in PVTL-1 Cell Line Newly Established from Acute Myeloid Leukemia Transformed from Polycythemia Vera. PLoS ONE 9:e84746, 2014.
更新日:2014年2月21日

概要
骨髄増殖性腫瘍(MPN)から進展した急性骨髄性白血病(AML)症例から、Jak2の活性化型変異(V617F)陽性の新規AML細胞株PVTL-1を樹立し解析した。Jak2-V617FはSTAT5を活性化する以外に、恒常的に活性化されたLynと供に、GSK3抑制とmTOR/4EBP1経路活性化をもたらす事で、生存維持と細胞増殖にそれぞれ必須の役割を果たしている事を見いだした。PVTL-1はMPNからAMLへ進展する機序を解明する上で、有用なモデルとなる事が期待される。
はじめに
細胞内チロシンキナーゼJak2は造血細胞におけるサイトカインシグナルにおいて根幹的な役割を担い、増殖やapoptosisの制御に極めて重要な位置を占める[1]。近年、骨髄増殖性腫瘍(MPN)ではJak2の活性化型点突然変異Jak2-V617Fが、大部分の症例で認められることが明らかとなった[2-4]。Jak2-V617Fを造血細胞株に導入すると、STAT5、PI3K/Akt、MEK/Erk経路などのJak2下流のシグナル経路が恒常的に活性化され、サイトカイン非依存性の自律的増殖をもたらし、また様々なマウスモデルでMPN様の表現型が再現できることから、病態形成に中心的な役割を果たすと考えられている[5]。一方、Srcファミリーチロシンキナーゼ(SFK)のLynは、やはり造血細胞の増殖や生存の制御に重要な役割を果たし、急性骨髄性白血病(AML)の病態や、慢性骨髄性白血病(CML)のチロシンキナーゼ阻害薬(TKI)治療への耐性獲得に重要な役割を果たすことが知られている[6]。
今回我々はMPNsの一種である真性多血症(PV)から骨髄異形成症候群(MDS)を経てAMLへと進展した症例からJak2-V617F陽性細胞株PVTL-1を樹立した。興味深いことにPVTL-1では、Jak2-V617FのみならずLynも恒常的に活性化しており、細胞の生存と増殖は両者からのシグナル伝達に依存していることが明らかとなった[7]。
症例とPVTL-1細胞株の樹立
症例は58歳時に真性多血症と診断され,10年間以上hydroxyurea内服にて治療を受けていたが、72歳時にMDSへと進展し、さらにその翌年にはAMLへと形質転換した。白血病細胞は5番および7番染色体の欠失を含む複雑な染色体異常を示し、Jak2-V617F変異が陽性と確認された。化学療法に抵抗性であり、AML診断後約4カ月で多臓器不全のため死亡した。AML診断時の末梢血の白血病細胞を分離し、白血病細胞株を樹立しPVTL-1と命名した。PVTL-1の細胞形態、細胞表面マーカー及び染色体核型は症例のものとほぼ一致し、Jak2-V617F変異アレル量の解析を行ったところ、ほぼ全てが変異アレルであり、Jak2-V617F変異ホモ接合体と考えられる。
Jak2-V617FとSFKによる内因性apoptosis経路の抑制と増殖誘導
PVTL-1はサイトカイン非依存性に増殖し、汎Jak阻害薬であるJak inhibitor-1(JI-1)にて用量依存性に増殖が抑制された。興味深いことに、CML等で発現する融合チロシンキナーゼBCR/ABLに対する第二世代TKIで、SFK阻害効果も有するdasatinib処理でもPVTL-1の増殖は用量依存性に著明に抑制された。一方、BCR/ABLに対する第一世代TKIのimatinib処理では増殖は影響を受けなかった。またJI-1およびdasatinibによる増殖抑制は相乗効果を認め、mitochondria膜電位変化、caspase-3活性化、annexin V細胞表面発現などのapoptosis解析により、JI-1やdasatinib処理はPVTL-1に mitochondria介在性の内因性apoptosis経路の活性化をもたらすことが示された。以上の結果より、PVTL-1細胞ではJak2-V617Fと供にSFKの活性化が内因性apoptosis経路の抑制と細胞増殖誘導に必須の役割を果たしているものと考えられる。
Jak2-V617Fと活性化したLynによる細胞内基質のリン酸化
PVTL-1では造血細胞における代表的SFKであるLynが強発現しており、活性化に特異的なY396の恒常的リン酸化を受けていたが、dasatinib処理にて著明な抑制を認めた。JakI-1とdasatinibによるそれぞれJak2-V617FとLyn抑制による効果の検討から、Jak2は主にSTAT3とSTAT5をリン酸化すると供にSHP2のリン酸化にも関与し、一方、恒常的に活性化したLynはSHP1, SHP2, Gab2, CrkLおよびc-Cbl等の多数の細胞内基質をリン酸化し、SHP2/Gab2やCrkL/c-Cbl等のシグナル複合体の形成に重要な役割を果たすことが明らかとなった。さらにLynはJak2の活性化特異的部位Y1007/1008以外のチロシンをリン酸化していることも見いだされ、Jak2-V617FとLyn活性化は相互に関連しつつPVTL-1の増殖や細胞機能の調節に影響を及ぼしていることが示唆された。
Jak2-V617FとLynからのGSK3抑制とmTOR活性化を介した生存と増殖のシグナル伝達機構
PVTL-1における細胞の生存と増殖制御のシグナル伝達経路の活性化を、Jak2-V617F陽性白血病細胞株HEL[3]やJak2-V617Fを発現させた造血細胞株[8]と比較検討した。活性化特異抗体を用いた検討で、PVTL-1ではErkとAktの活性化は認めず、HELでは前者のみJak2-V617F依存性に活性化していた(図1)。一方で、これらの下流で活性化され蛋白合成や細胞増殖に重要な役割を果たすmTOR経路[9]に関しては、mTOR、p70S6K、S6RP, 4EBP1等の恒常的なリン酸化を両者の細胞で認めた。これらのリン酸化は、HELではJak2-V617F依存性、PVTL-1ではJak2-V617FとLynの両者に依存性に生じている事が、阻害薬を用いた検討で明らかになった(図1)。また、一般的にAktによりリン酸化を受け機能が抑制され、apoptosis制御に関与するGSK3[10]に関しても検討したが、両細胞株ともに恒常的なリン酸化を認め、HELではJak2-V617F依存性、PVTL-1ではJak2-V617FとLynの両者に依存性であった。尚、PVTL-1においてGSK3のリン酸化は、Akt阻害薬MK-2206では抑制されなかったが、PI3K阻害薬GDC-0941では抑制されたことから、PI3K下流でAkt非依存性に生じているものと考えられる。GSK3はapoptosisの制御に関与することが知られており[10]、その機能を評価するためGSK3阻害薬SB216763処理で検討を行ったところ、Lyn阻害によるcaspase活性化とapoptosis誘導は有意に抑制されたが、Jak2-V617F阻害によるapoptosis誘導には影響を認めなかった(図1)。
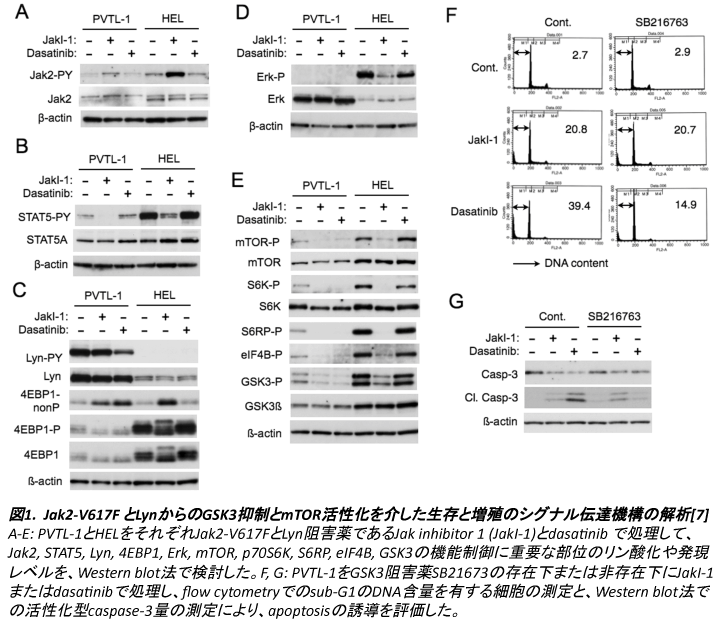
考察
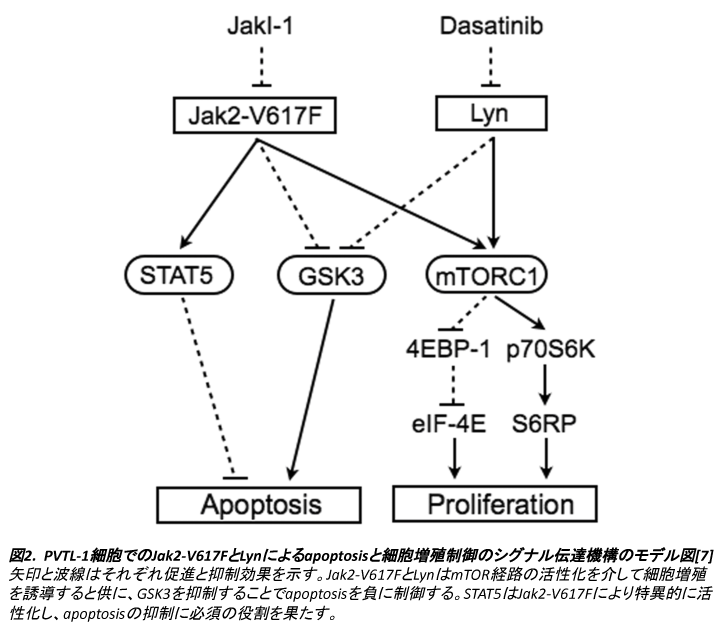
MPNからAMLに進展した症例から樹立したPVTL-1細胞株では、MPN発症に関与するJak2のV617F変異による活性化のみでなく、Lynの強発現と恒常的活性化を認め、両者が細胞の生存と増殖に必須の役割を果たしていることが見出された。Jak2-V617Fは特異的にSTAT5の活性化を担っていることが確認されたが、それ以外にAktの活性化を介さずに、少なくとも一部はPI3K依存性に、GSK3の抑制とmTOR経路の活性化を誘導していることが明らかとなった(図2)。活性化したLynも同様にGSK3抑制とmTOR系の活性化に必須の役割を果たし、また、Jak2のY1007/1008以外の部位のチロシンリン酸化をもたらした事から、Jak2-V617Fの活性制御にも何らかの影響を持つ事も予想される。Jak2-V617FとLynはPVTL-1細胞において、主にmTOR経路の活性化を介して細胞増殖を誘導し、GSK3の抑制を介して内因性のapoptosis誘導機構を抑制するものと考えられるが、GSK3阻害のみではJakI-1によるapoptosis誘導は回避出来なかった事から、Jak2-V617FによるSTAT5の活性化もやはりapoptosisの抑制には必須の役割を果たすものと考えられる(図2)。
PVTL-1細胞株とその細胞内シグナルの解析結果は、MPNの病態解明のみでなく、MDSやAMLへの進展機構の解明や、これらの難治性造血器腫瘍に対する新たな分子標的療法開発へ向けての研究にも寄与することが期待される。
参考文献
1. Ihle JN (1995) Cytokine receptor signalling. Nature 377: 591-594.
2. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, et al. (2005) A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 352: 1779-1790.
3. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, et al. (2005) Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7: 387-397.
4. Tefferi A (2012) Polycythemia vera and essential thrombocythemia: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 87: 285-293.
5. Quintas-Cardama A (2013) The role of Janus kinase 2 (JAK2) in myeloproliferative neoplasms: therapeutic implications. Leuk Res 37: 465-472.
6. Ingley E (2012) Functions of the Lyn tyrosine kinase in health and disease. Cell Commun Signal 10: 21.
7. Nagao T, Kurosu T, Umezawa Y, Nogami A, Oshikawa G, et al. (2014) Proliferation and Survival Signaling from Both Jak2-V617F and Lyn Involving GSK3 and mTOR/p70S6K/4EBP1 in PVTL-1 Cell Line Newly Established from Acute Myeloid Leukemia Transformed from Polycythemia Vera. PLoS One 9: e84746.
8. Nagao T, Oshikawa G, Wu N, Kurosu T, Miura O (2011) DNA damage stress and inhibition of Jak2-V617F cause its degradation and synergistically induce apoptosis through activation of GSK3beta. PLoS One 6: e27397.
9. Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149: 274-293.
10. Chiara F, Rasola A (2013) GSK-3 and mitochondria in cancer cells. Front Oncol 3: 16.